Functional annotation and importance of marine bacterial transporters of plankton exometabolites
William F. Schroer, Hannah E. Kepner, Mario Uchimiya, Catalina Mejia, Lidimarie Trujillo Rodriguez, Christopher R. Reisch & Mary Ann Moran
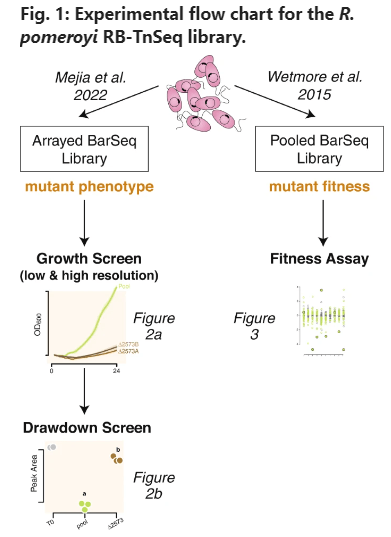
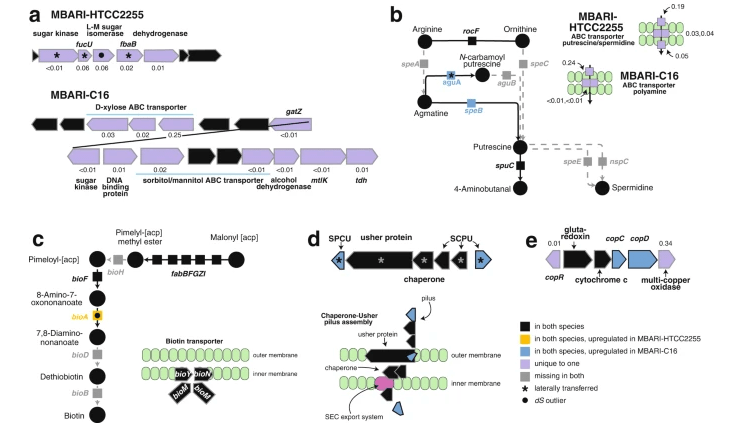
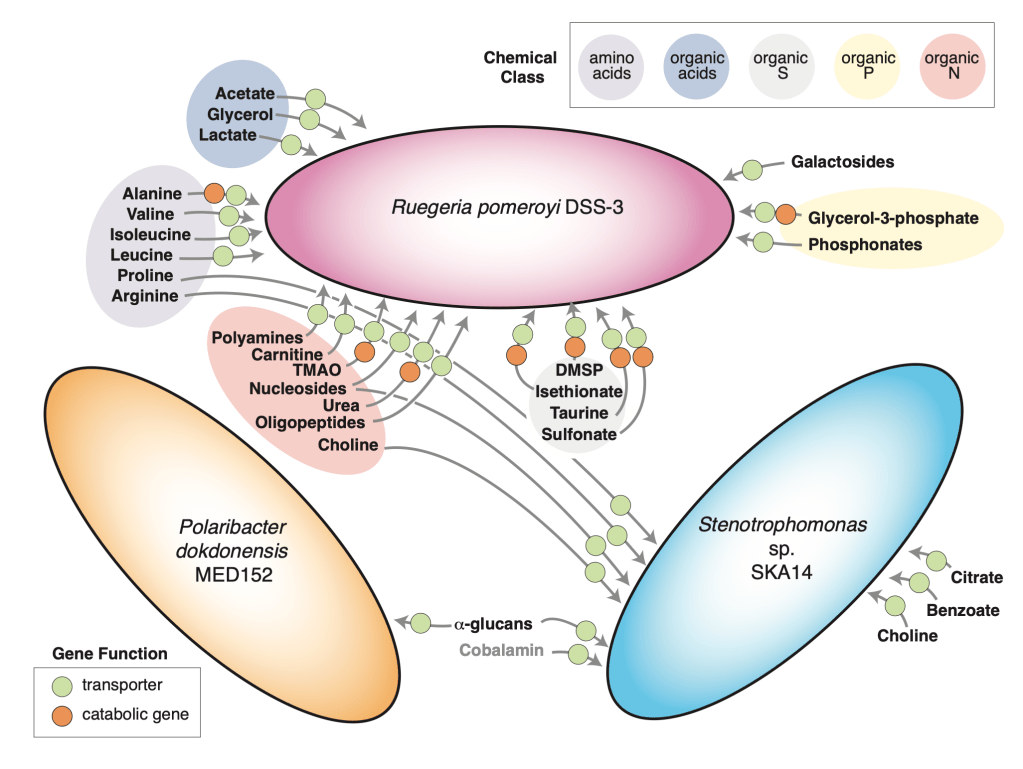
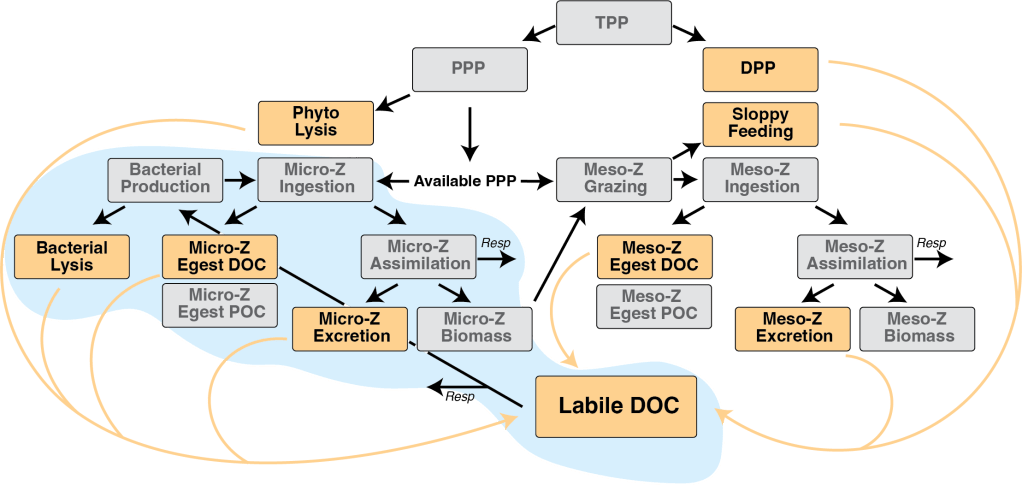
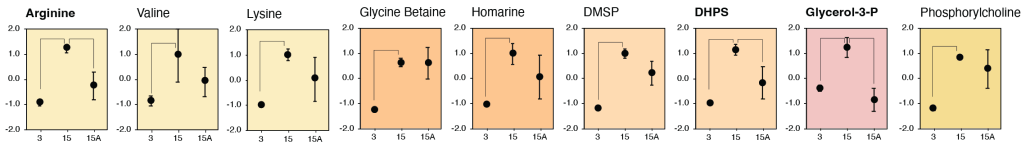
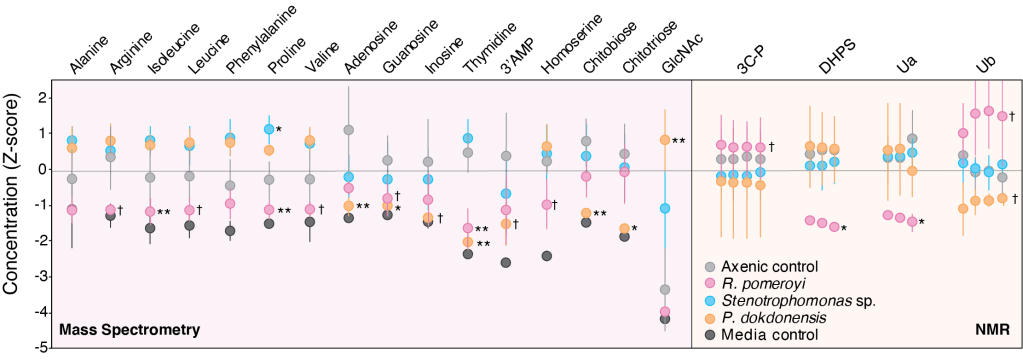
Metabolite exchange within marine microbial communities transfers carbon and other major elements through global cycles and forms the basis of microbial interactions. Yet lack of gene annotations and concern about the quality of existing ones remain major impediments to revealing currencies of carbon flux. We employed an arrayed mutant library of the marine bacterium Ruegeria pomeroyi DSS-3 to experimentally annotate substrates of organic compound transporter systems, using mutant growth and compound drawdown analyses to link transporters to their cognate substrates. Mutant experiments verified substrates for thirteen R. pomeroyi transporters. Four were previously hypothesized based on gene expression data (taurine, glucose/xylose, isethionate, and cadaverine/putrescine/spermidine); five were previously hypothesized based on homology to experimentally annotated transporters in other bacteria (citrate, glycerol, N-acetylglucosamine, fumarate/malate/succinate, and dimethylsulfoniopropionate); and four had no previous annotations (thymidine, carnitine, cysteate, and 3-hydroxybutyrate). These bring the total number of experimentally-verified organic carbon influx transporters to 18 of 126 in the R. pomeroyi genome. In a longitudinal study of a coastal phytoplankton bloom, expression patterns of the experimentally annotated transporters linked them to different stages of the bloom, and also led to the hypothesis that citrate and 3-hydroxybutyrate were among the most highly available bacterial substrates. Improved functional annotation of the gatekeepers of organic carbon uptake is critical for deciphering carbon flux and fate in microbial ecosystems.